Introduction
α-synuclein, a protein predominantly found in presynaptic nerve terminals and nuclei, has captivated researchers for its critical roles in neuronal health and its intriguing structural properties. First identified in the electric ray Torpedo marmorata, its name reflects its localization in the synapse and nucleus.
The Synuclein Gene Family
Encoded by the SNCA, SNCB, and SNCG genes, α-synuclein belongs to a family of proteins essential for neuronal function. These genes encode alpha-, beta-, and gamma-synuclein, which share approximately 60% sequence homology at the amino acid level, particularly in the N-terminal region where the KTKEGV repeat motifs are conserved. This homology supports their shared roles in synaptic function and lipid membrane interactions. Notably, mutations in SNCA, such as A53T, E46K, and A30P, are associated with protein misfolding and aggregation.
Structural Insights into Alpha-Synuclein
1. Domains and Features:
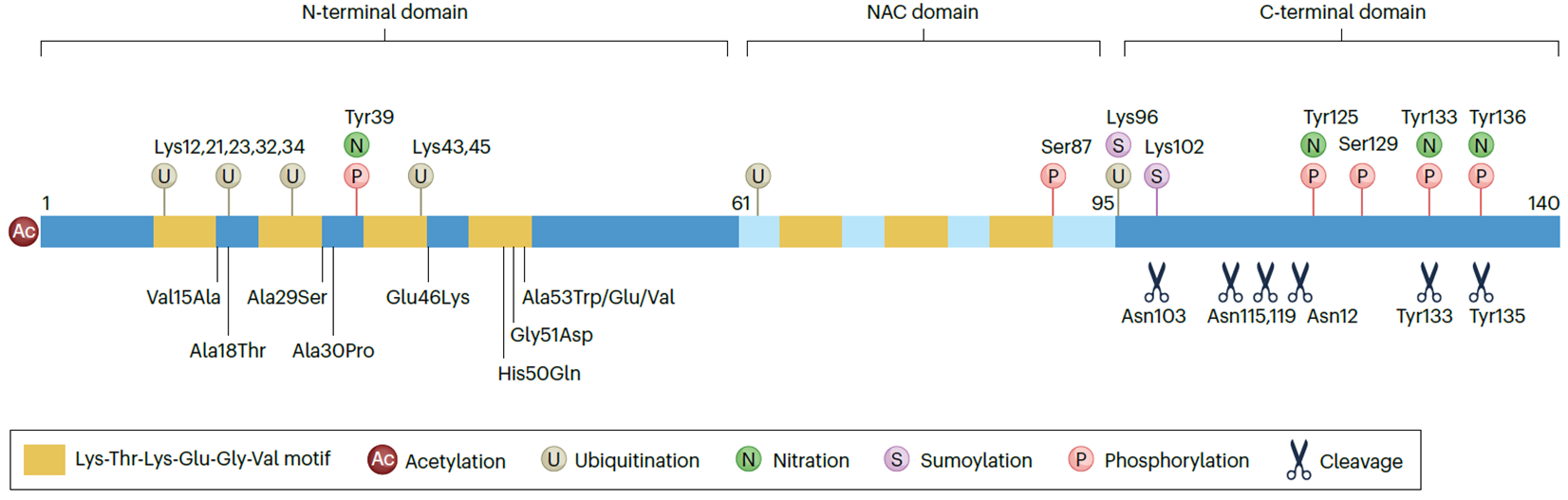
- N-terminal Domain (Residues 1–60): Facilitates membrane binding and lipid interactions through its amphipathic alpha-helical regions.
- NAC Region (Residues 61–95): A hydrophobic core prone to forming beta-sheet structures, critical for aggregation and amyloid fibril formation.
- C-terminal Domain (Residues 96–140): Acidic and highly charged, this region interacts with metal ions (e.g., calcium, copper) and modulates solubility and aggregation.
2. Repeat Motifs:
- Contains KTKEGV motifs essential for lipid binding, self-assembly, and regulating vesicle dynamics.
Each amino acid in the KTKEGV motif contributes specific properties:
K (Lysine): Positively charged, enhances interaction with negatively charged lipid membranes.
T (Threonine): Polar and hydrophilic, adds stability through potential hydrogen bonding.
K (Lysine): Repeated lysine residues reinforce membrane association via electrostatic attraction.
E (Glutamate): Negatively charged, balances the amphipathic nature and may participate in ionic interactions.
G (Glycine): Small and flexible, promotes structural adaptability in the motif.
V (Valine): Hydrophobic, contributes to the motif's ability to interact with lipid bilayers.
This combination of residues allows the KTKEGV motif to mediate alpha-helical formation, stabilize protein-membrane interactions, and facilitate vesicle clustering.
3. Dynamic Nature:
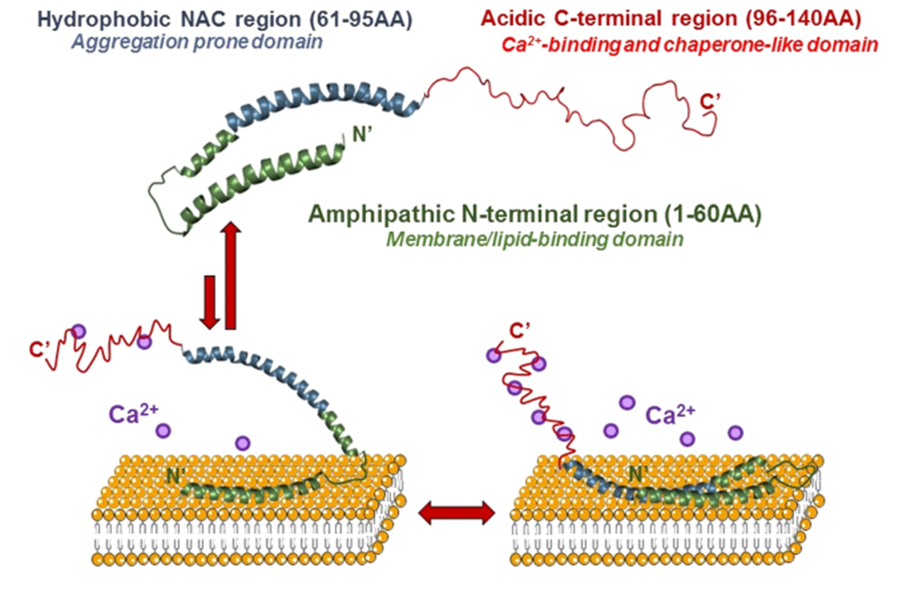
- In its native state, α-synuclein is intrinsically disordered, which allows it to remain highly flexible and interact with a wide variety of cellular components. This disordered nature enhances its ability to adapt to changing cellular environments and engage in diverse biological processes.
- Upon binding to lipid membranes, α-synuclein transitions into an alpha-helical structure. This structural shift is driven by the protein’s amphipathic N-terminal domain, which aligns with lipid bilayers, stabilizing the protein-membrane interaction. This dynamic transition is critical for its physiological roles, such as vesicle clustering and membrane remodeling.
- The structural flexibility of α-synuclein enables it to exist in multiple conformational states, including monomers, oligomers, dimers, tetramers, and fibrils. These states are influenced by environmental factors such as pH, ionic strength, and lipid composition.
- Monomer Stabilization: Heat shock proteins (HSPs), particularly Hsp70 and Hsp90, play a crucial role in stabilizing α-synuclein monomers. These molecular chaperones prevent the misfolding and aggregation of α-synuclein by binding to its hydrophobic regions, particularly in the NAC domain. This interaction not only maintains the functional state of monomers but also reduces the pool of aggregation-prone species, providing a protective mechanism against synucleinopathies.
- Dimers and Tetramers: Native α-synuclein is thought to exist predominantly as a helical tetramer under physiological conditions, providing stability and reducing aggregation-prone monomers. These smaller oligomeric forms, while generally considered stable, differ from larger, less-structured oligomers often implicated in toxicity. The disruption of tetramers, often by mutations or environmental stress, increases the cytosolic pool of monomers, leading to dimer and larger oligomer formation. While tetramers may serve protective roles, dimers are transitional states that can facilitate aggregation into toxic forms, thereby playing a key role in the aggregation cascade.
- While this adaptability underpins its physiological functionality, it also predisposes α-synuclein to pathological misfolding. Misfolding events often involve the NAC region, which forms beta-sheet-rich cores during fibril formation, contributing to aggregation and toxicity.
- Understanding these dynamic transitions provides insight into how α-synuclein balances its roles in normal cellular activity with its potential for pathological aggregation.
Physiological Functions
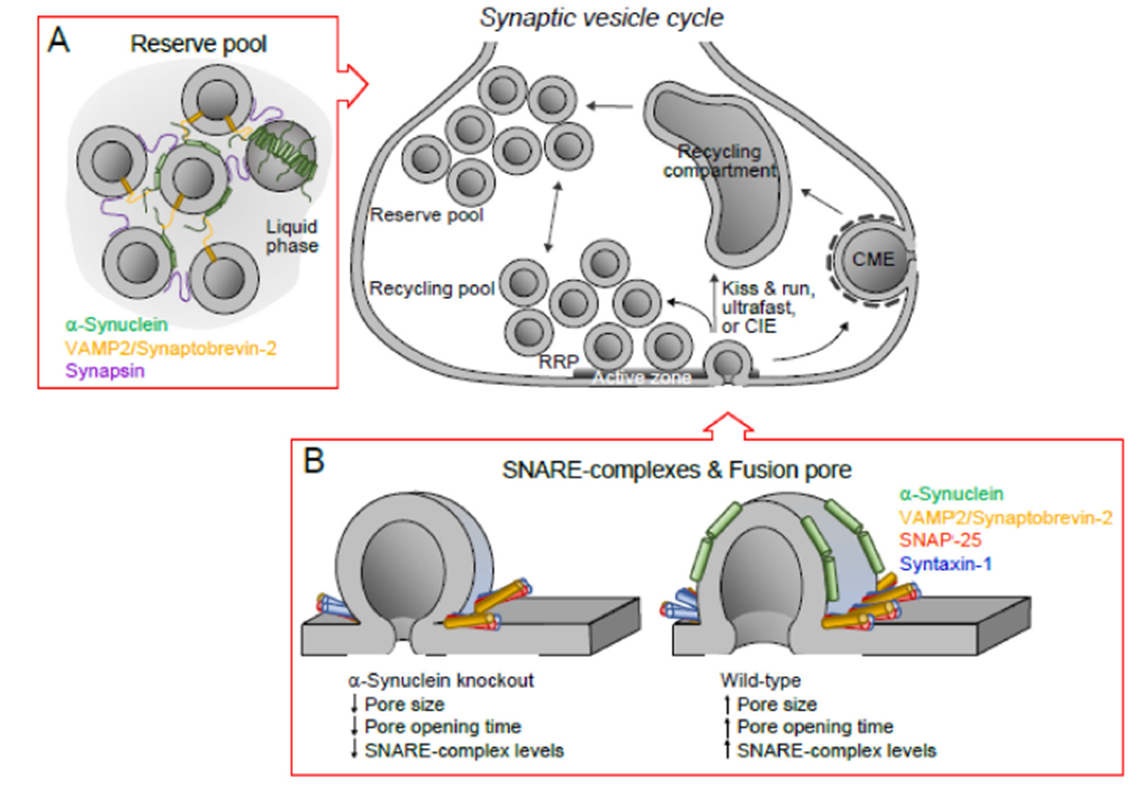
- Synaptic Vesicle Regulation: α-synuclein plays a crucial role in clustering synaptic vesicles and facilitating neurotransmitter release. Studies using knockout models reveal significant deficits in neurotransmission without this protein. Known interacting partners include SNARE complex proteins such as VAMP2 (synaptobrevin-2) and syntaxin-1, which are critical for vesicle docking and fusion. Additionally, α-synuclein interacts with Rab3a, a GTPase essential for vesicle trafficking, and phospholipase D2, influencing vesicle lipid composition.
- Membrane Interaction:
The protein stabilizes vesicle dynamics and influences lipid bilayer curvature. Its preference for negatively charged lipid environments such as phosphatidylserine-rich membranes underscores its specialized synaptic functions. This preference for curved lipid membranes arises from the amphipathic alpha-helical regions in the N-terminal domain, which adapt to the curvature, reducing the energy barrier for binding. Curved membranes, often found in synaptic vesicles, offer an optimal environment for α-synuclein to stabilize vesicle structures and promote clustering.
Toxicity of Oligomers and Fibrils
α-synuclein's ability to aggregate into oligomers and fibrils represents a double-edged sword in neurobiology. While necessary for some cellular processes, these aggregates can exert significant toxicity:
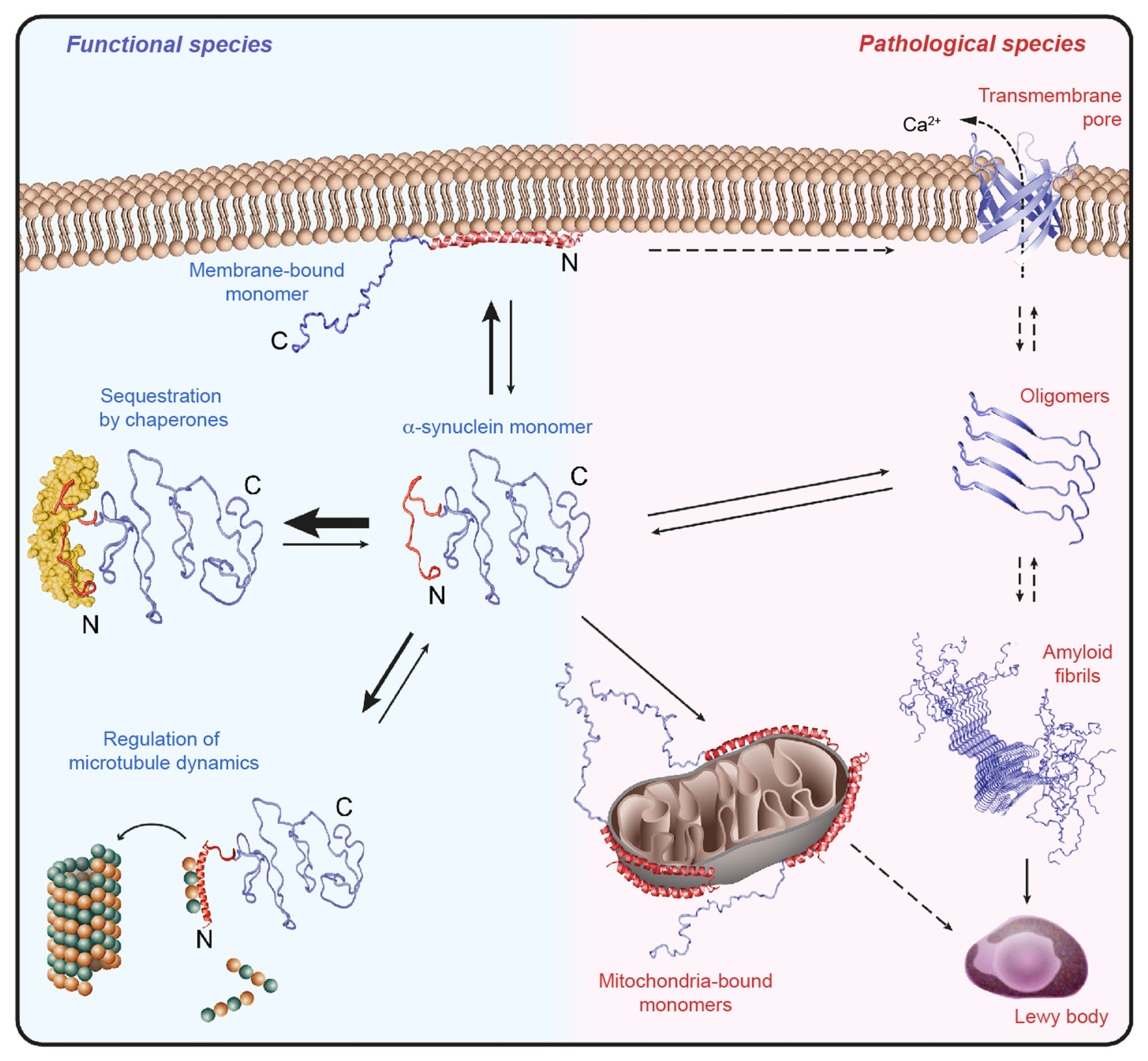
1. Oligomeric Toxicity:
- Oligomers are considered the most neurotoxic form of α-synuclein. They can interact with lipid membranes, forming pore-like structures that disrupt membrane integrity.
- This disruption leads to ion dysregulation, particularly calcium influx, which can trigger apoptotic pathways.
- Oligomers impair synaptic vesicle recycling and neurotransmitter release, destabilizing neuronal communication.
- Additionally, oligomers directly affect mitochondrial function by disrupting mitochondrial membranes. This impairs ATP production, increases oxidative stress, and initiates mitochondrial-dependent apoptosis.
- Oligomers are considered the most neurotoxic form of alpha-synuclein. They can interact with lipid membranes, forming pore-like structures that disrupt membrane integrity.
- This disruption leads to ion dysregulation, particularly calcium influx, which can trigger apoptotic pathways.
- Oligomers impair synaptic vesicle recycling and neurotransmitter release, destabilizing neuronal communication.
2. Fibril-Associated Toxicity:
- Aggregated fibrils also affect mitochondrial integrity, impairing mitochondrial membranes and disrupting energy metabolism.
- This mitochondrial dysfunction leads to increased oxidative stress and reduced ATP production, exacerbating neuronal vulnerability.
- Mature fibrils, though less toxic than oligomers, serve as a structural core for the formation of Lewy Bodies, a hallmark of synucleinopathies.
- Mature fibrils, though less toxic than oligomers, serve as a structural core for the formation of Lewy Bodies, a hallmark of synucleinopathies.
- Fibrils act as seeds for further aggregation, spreading pathology through a prion-like mechanism.
- They overwhelm cellular proteostasis mechanisms, including the ubiquitin-proteasome system and autophagy, leading to cellular stress and neurodegeneration.
Understanding the balance between physiological aggregation and pathological toxicity is critical for developing therapeutic interventions targeting alpha-synuclein aggregation.
Structural Polymorphism
Alpha-synuclein exhibits structural polymorphism that affects its function and aggregation propensity. This polymorphism refers to the ability of alpha-synuclein to form different structural conformations depending on various factors:
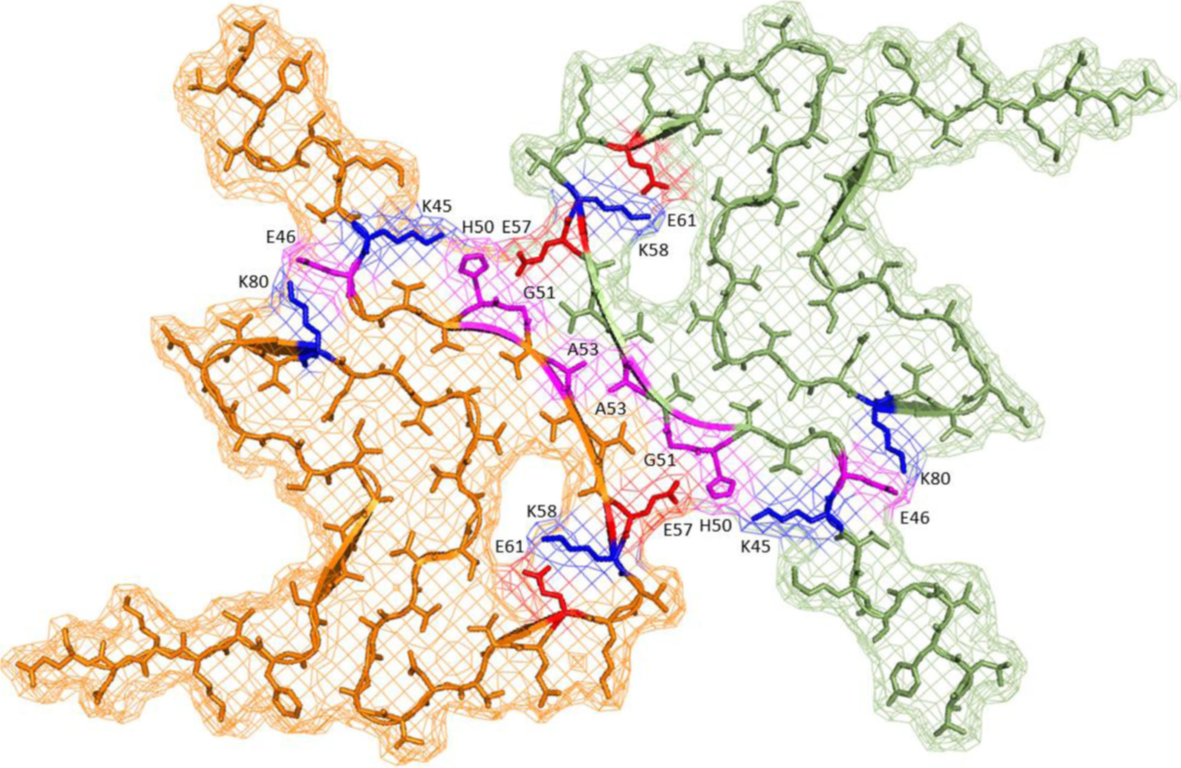
- Beta-Sheet Core in Fibrils:
Aggregated forms exhibit a beta-sheet-rich core, stabilized by hydrophobic and electrostatic interactions, primarily within residues 61–95 (NAC region). These fibril cores are further stabilized by salt bridges, which involve ionic interactions between charged residues, and a hydrophobic zipper, where hydrophobic side chains interlock to reduce exposure to the aqueous environment. These molecular features contribute to the packing density and stability of the fibril structures, leading to distinct structural states. Among these, two dominant fibril polymorphs have been identified:
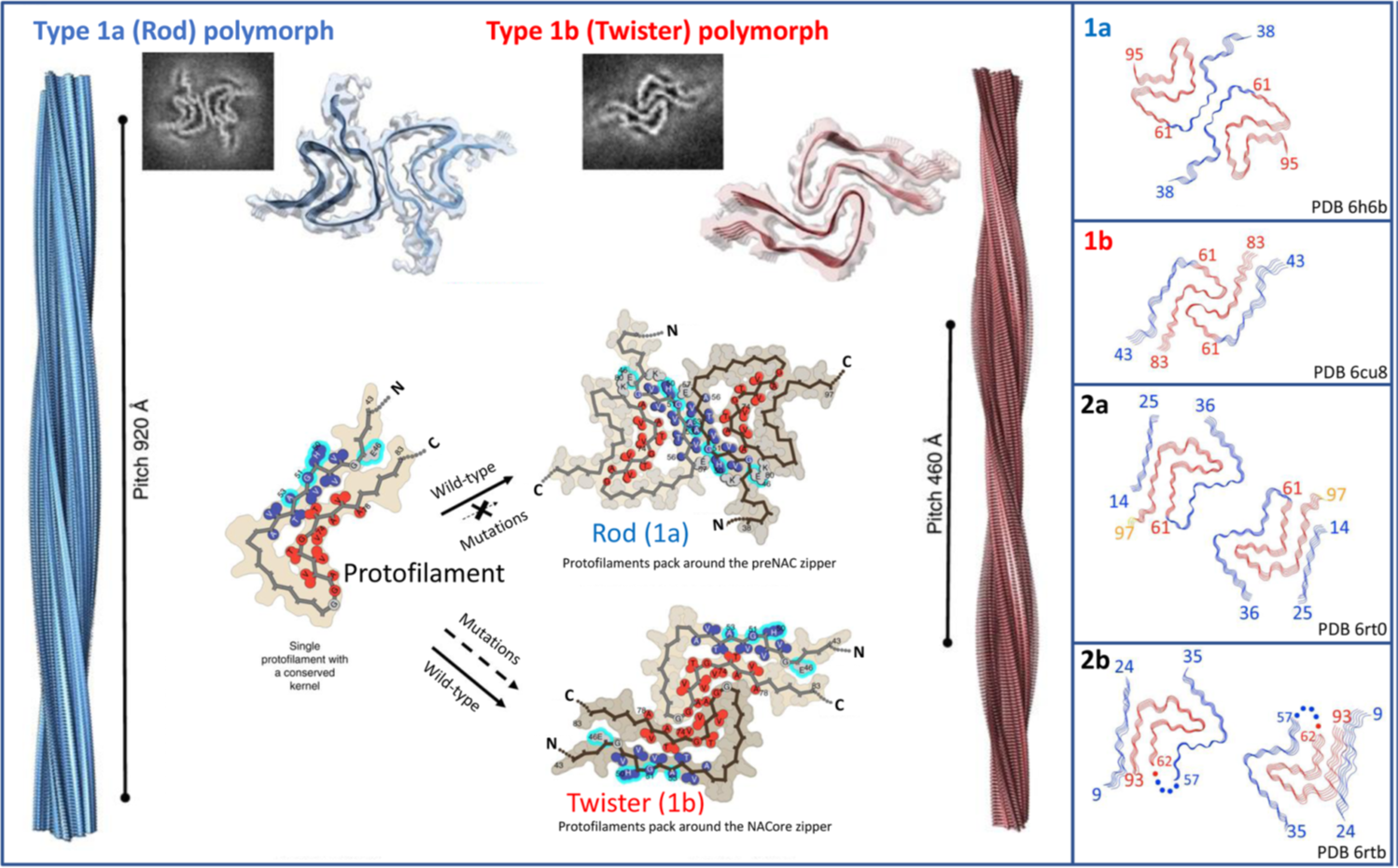
- Rod Form: Characterized by elongated, straight fibril structures with uniform packing. Rod fibrils are associated with slower propagation rates and moderate toxicity. These fibrils are generally more stable due to consistent hydrogen bonding and lower conformational strain within the beta-sheet core.
- Twister Form: Features twisted, helical fibrils with more complex packing arrangements. These are linked to higher aggregation rates, greater structural flexibility, and increased toxicity. The twisting of fibrils introduces structural instability, making them more prone to fragmentation, which can facilitate the prion-like propagation observed in Parkinson’s Disease. Unlike other fibril-related pathologies, such as amyloid-beta in Alzheimer’s Disease, α-synuclein fibrils exhibit a higher propensity for intercellular spread due to their unique interaction with cellular membranes. This specificity may explain the characteristic progression patterns observed in synucleinopathies.
Both fibril forms, rod and twister, have been identified in post-mortem analyses of Parkinson’s Disease patient brains. Their presence underscores the heterogeneity of α-synuclein aggregation in vivo, with rod forms contributing to slower disease progression and twister forms being associated with more aggressive pathological outcomes.
- Impact of Mutations:
Genetic mutations, such as A53T (residue 53), E46K (residue 46), and A30P (residue 30), induce significant changes in fibril architecture. For instance, A53T mutations enhance aggregation rates and promote the formation of fibrils with greater structural stability, while E46K influences beta-sheet stacking interactions, particularly in the NAC region.
- Environmental Influences:
Environmental conditions, including pH, ionic strength, and metal ion concentrations, dramatically alter fibril morphology. For example, high concentrations of calcium or copper ions interact with residues in the C-terminal domain (96–140) and can accelerate aggregation, inducing fibril polymorphs with unique toxic profiles.
- Post-Translational Modifications (PTMs):
Modifications such as phosphorylation (e.g., at residue S129), acetylation, and truncation at specific residues can shift the aggregation pathway, favoring certain fibril strains over others. These PTMs not only affect structural stability but also influence the biological activity and cellular toxicity of the fibrils, particularly within the NAC and C-terminal regions.
Clinical Relevance
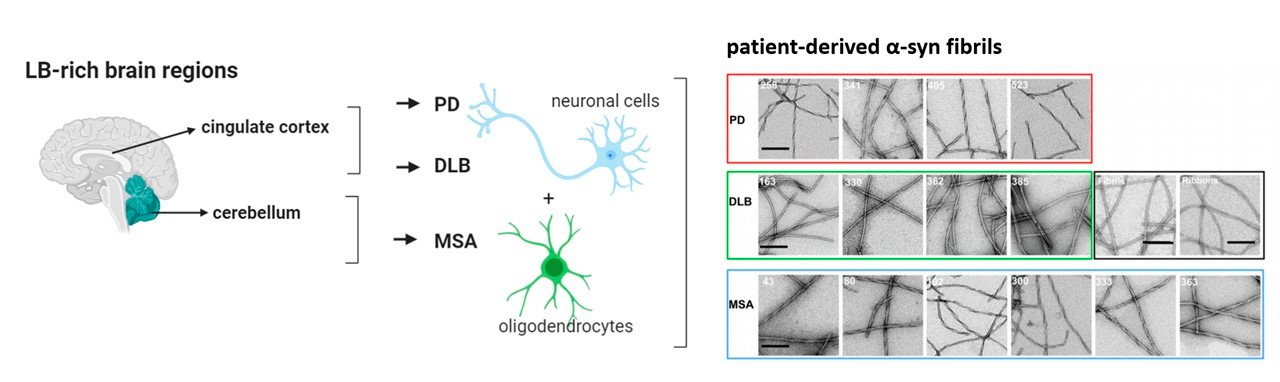
Structural polymorphism underlies the clinical diversity observed in synucleinopathies. For instance:
- Parkinson’s Disease (PD): Characterized by the presence of α-synuclein fibrils in neurons, particularly in dopaminergic neurons of the substantia nigra. Among fibril polymorphisms, rod fibrils are linked to slower disease progression due to their stability and reduced fragmentation. In contrast, twister fibrils, which are more prone to fragmentation and propagation, correlate with more severe motor symptoms and rapid disease progression. These differences underscore the pathological impact of fibril structures.
- Multiple System Atrophy (MSA): Exhibits distinct α-synuclein fibril conformations in oligodendrocytes, forming glial cytoplasmic inclusions. Fibrils in MSA are typically shorter and more fragmented than those found in PD, with structural features that enhance seeding activity and toxicity. These shorter fibrils exhibit increased surface area, which facilitates rapid interaction with cellular components and accelerates seeding of new aggregates. This higher reactivity correlates with the aggressive progression and widespread neurodegeneration observed in MSA, particularly in the cerebellum and basal ganglia. These fibrils contribute to the more aggressive progression of MSA compared to PD and are associated with widespread neurodegeneration in the cerebellum and basal ganglia.
- Dementia with Lewy Bodies (DLB): Features widespread cortical α-synuclein fibril deposition, correlating with severe cognitive decline and hallucinations. In DLB, the fibrils often exhibit polymorphisms distinct from those in PD or MSA, with characteristics that enable cortical spread and higher neurotoxicity. These fibrils show a high degree of flexibility and interaction with cortical structures, facilitating their widespread distribution. Additionally, they are associated with significant synaptic disruption, contributing to the hallmark cognitive decline and hallucinations observed in DLB. Their unique properties highlight the role of fibril polymorphism in disease-specific pathology. These fibrils contribute to the distinct clinical and pathological profiles of DLB.
Understanding these polymorphisms provides insights into disease-specific mechanisms of neurodegeneration and paves the way for targeted therapeutic interventions. For instance, therapies aimed at stabilizing less toxic fibril forms or preventing prion-like propagation could significantly alter disease outcomes.
Conclusion
Alpha-synuclein's structural dynamics hold the key to understanding its physiological and pathological roles. Its intrinsically disordered nature, combined with its ability to adopt various conformations, makes it a fascinating model for studying protein aggregation and cellular interactions. Unraveling the complexities of its structure will pave the way for advances in diagnostics and therapeutics.
References
1. Fauvet, B., Mbefo, M. K., Fares, M.-B., et al. (2012). α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and *Escherichia coli* exists predominantly as disordered monomer. *Journal of Biological Chemistry*, 287(19), 15345–15364. [https://doi.org/10.1074/jbc.M111.318949](https://doi.org/10.1074/jbc.M111.318949)
2. Ulmer, T. S., Bax, A., Cole, N. B., & Nussbaum, R. L. (2005). Structure and dynamics of micelle-bound human alpha-synuclein. *Journal of Biological Chemistry*, 280(10), 9595–9603. [https://doi.org/10.1074/jbc.M411805200](https://doi.org/10.1074/jbc.M411805200)
3. Lashuel, H. A., Petre, B. M., Wall, J., et al. (2002). Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. *Journal of Molecular Biology*, 322(5), 1089–1102. [https://doi.org/10.1016/S0022-2836(02)00813-2](https://doi.org/10.1016/S0022-2836\(02\)00813-2)
4. Breydo, L., Wu, J. W., & Uversky, V. N. (2012). Alpha-synuclein misfolding and Parkinson's disease. *Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease*, 1822(2), 261–285. [https://doi.org/10.1016/j.bbadis.2011.10.002](https://doi.org/10.1016/j.bbadis.2011.10.002)
5. Stefanis, L. (2012). Alpha-synuclein in Parkinson’s disease: From pathology to therapeutic target. *Molecular Neurobiology*, 47(2), 529–539. [https://doi.org/10.1007/s12035-012-8280-y](https://doi.org/10.1007/s12035-012-8280-y)
6. Hyejin Park, Tae-In Kam, Valina L Dawson, Ted M Dawson (2024). Alpha-synuclein pathology as a target in neurodegenerative diseases. Nat Rev Neurol, 2024 Nov 28. doi: 10.1038/s41582-024-01043-w.